Hoe AI het zoeken naar medicijnen slimmer maakt
Francesca Grisoni en haar collega's gebruiken AI-taalmodellen om een revolutie te ontketenen in de manier waarop we medicijnen ontwikkelen.

Deep learning heeft de wereld van de kunstmatige intelligentie radicaal veranderd. Nu zorgen neurale netwerken voor een revolutie in de manier waarop we nieuwe geneesmiddelen ontwerpen. We praten met Francesca Grisoni, universitair docent aan de faculteit Biomedical Engineering, die voorop loopt in het onderzoek naar het automatiseren van de ontwikkeling van medicijnen.
Laten we bij het begin beginnen. De ontwikkeling van geneesmiddelen is, zoals bekend, een extreem moeizaam proces. Het aantal moleculen dat kan dienen als uitgangspunt voor een geneesmiddel, ligt naar schatting in de orde van 1060 - 10100, veel meer dan er sterren zijn in het waarneembare heelal. Een molecuul vinden dat precies de eigenschappen heeft die je wenst voor je geneesmiddel, is dan ook zoiets als het vinden van een naald in een hooiberg.
"De meeste farmaceutische moleculen worden ontworpen om specifieke doeleiwitten in onze cellen te moduleren. Door interactie met deze doelwitten kan een geneesmiddel de activiteit van dat eiwit afremmen of bevorderen. Dit effect, bekend als bioactiviteit, kan worden gebruikt om ziekten te genezen of te voorkomen", legt Grisoni uit.

"Het centrale idee bij het ontwerpen van geneesmiddelen is dat je moleculen wilt identificeren die wel actief zijn voor de beoogde doelwitten, maar niet voor andere doelwitten die ongewenste bijwerkingen kunnen veroorzaken. Gezien het grote aantal mogelijke moleculen dat in aanmerking komt, is het echt heel lastig om efficiënt door deze 'chemische ruimte' te navigeren.”
En dat is nog maar het begin. Zodra je een potentiële kandidaat hebt gevonden voor je medicijn, moet je het molecuul (of de verbinding) nog in een laboratorium produceren (een proces dat bekend staat als chemische synthese), en in steeds complexere experimenten testen. Dit is niet alleen een zeer tijdrovend karwei (veel moleculen lopen uit op een mislukking), maar ook zeer duur. Dit is een van de redenen waarom de grote farmaceutische bedrijven zo angstvallig hun zwaarbevochten octrooien bewaken.
Medicijnen ontwikkelen met computers
Het zal dan ook niet verbazen dat medicinale chemici en biologen zo'n 30 jaar geleden een beroep deden op computers om het ontdekkings- en ontwikkelingsproces van geneesmiddelen te versnellen. "Computers kunnen helpen bij het selecteren van de moleculen die het meest waarschijnlijk effectief zijn voor het beoogde doel en die kunnen worden gesynthetiseerd”, aldus Grisoni.
"Eén manier om dat te doen is wat wij virtuele screening noemen, waarbij je computationele methoden gebruikt om te bepalen welke kandidaten je wil testen uit een bibliotheek met moleculen waarvan je weet dat ze kunnen worden gesynthetiseerd. Deze bibliotheken zijn meestal veel kleiner dan het hele chemische universum (tussen 103 en 106 moleculen), dus veel gemakkelijker te doorzoeken. In sommige gevallen wil je echter andere regio's van de chemische ruimte verkennen, die niet in dergelijke virtuele screeningbibliotheken zijn opgenomen."
Hier kan 'de novo'-design, waarbij je moleculen ‘vanuit het niets’ ontwerpt, uitkomst bieden. Grisoni: "De novo-design heeft als bijkomend voordeel dat je moleculen kunt genereren die precies gericht zijn op de doelen die je wilt bereiken, en hopelijk ook moleculen waar nog niemand aan heeft gedacht."
Van ontwerpen met regels naar deep learning
Maar hoe bouw je zulke moleculen van de grond af op? Tot voor kort gebeurde dit meestal door het volgen van specifieke regels. "Denk aan hoe grammatica werkt in taal. Als je een heleboel woorden willekeurig achter elkaar zet, krijg je een zin die nergens op slaat. Dus heb je regels nodig. Op dezelfde manier kun je regels bedenken over hoe je atomen of moleculaire fragmenten samenvoegt tot een verbinding die niet alleen chemisch zin heeft, maar ook de gewenste biologische eigenschappen heeft."
Maar net als de computerlinguïsten die in de jaren '80 en '90 van de vorige eeuw tevergeefs probeerden vertalingen te automatiseren door middel van een groot regelboek, zou de op regels gebaseerde aanpak al snel tegen zijn grenzen kunnen aanlopen.
"In sommige gevallen realiseer je je al snel dat de regels ofwel te beperkend zijn, ofwel te complex”, zegt Grisoni. Het is hier dat machine learning, en meer specifiek deep learning, te hulp schiet. Het heeft onderzoekers zoals Grisoni in staat gesteld om niet alleen automatisch de 'grammatica' van bekende verbindingen te leren (welke elementen zijn nodig om een geldige molecule te maken die ook kan worden gesynthetiseerd?), maar ook de 'semantiek' ervan (welke elementen zijn nodig om de gewenste bio-activiteit op een bepaald doelwit te hebben?).
Lachende moleculen
Om dit voor elkaar te krijgen, gebruiken de onderzoekers deep learning-modellen die ontleend zijn aan Natural Language Processing, dezelfde techniek die ook een revolutie teweeg heeft gebracht in de wereld van automatische vertalingen en spraakherkenning (en waar we onmisbare apps als Google Translate en Siri aan te danken hebben). Om NLP te kunnen gebruiken bij het ontwerpen van geneesmiddelen, moest de structuur van de moleculen eerst worden uitgedrukt als een reeks woorden.
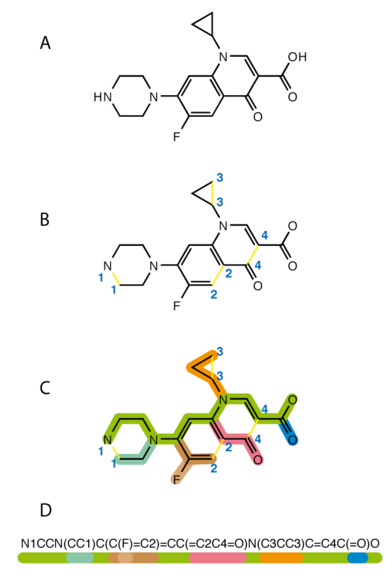
Gelukkig is er sinds de jaren tachtig zo'n taal beschikbaar: SMILES (zie afbeelding). "Door per keer één karakter toe te voegen om de SMILES-keten af te maken, is het NLP-model in staat automatisch nieuwe moleculen te genereren. Dit proces is niet willekeurig. De nieuwe tekens worden gekozen op basis van wat het model heeft geleerd van eerder beschikbare gegevens", legt Grisoni uit. "Vergelijk het met Google Search, dat automatisch je zoekopdracht aanvult op basis van eerdere zoekopdrachten."
Maar anders dan Google Search of Google Translate worden Grisoni en haar collega's geconfronteerd met een lastig probleem dat specifiek is voor de wereld van medicijnontwikkeling: het gebrek aan data om de computeralgoritmen die worden gebruikt om nieuwe moleculen te genereren, te trainen. "Grote datasets voor deep learning in geneesmiddelenontwerp zijn vrij schaars. In sommige gevallen heb je maar een handvol verbindingen waarvan bekend is dat ze werken op een bepaald doelwit," legt ze uit.
Omgaan met schaarse data
Het was een van de noten die ze moest kraken voor een publicatie over generatieve kunstmatige intelligentie, dat ze samen met oud-collega's van ETH Zürich schreef en dat onlangs werd gepubliceerd in het tijdschrift Science Advances. In het artikel combineren de onderzoekers voor het eerst een ‘regelvrije’ deep learning-aanpak om bioactieve moleculen te genereren met on-chip-synthese, een vorm van geautomatiseerde synthese op miniatuurschaal die de hoeveelheid menselijke arbeid verder minimaliseert.

Om het datatekort te omzeilen, gebruikten de onderzoekers een methode die transfer learning wordt genoemd.
"Het basisidee is dat je gebruik maakt van gegevens die op de een of andere manier verband houden met je probleem, maar waarvoor veel meer voorbeelden beschikbaar zijn, ook al zijn het niet precies de gegevens die je nodig hebt. Denk aan wanneer iemand voor de eerste keer een wetenschappelijk artikel schrijft. Misschien heeft hij voordien 50 gelijkaardige documenten gelezen, en dan is hij al in staat om het zijne te beginnen schrijven. Maar ze zijn natuurlijk niet vanaf nul begonnen, ze hebben hun hele leven leren lezen en schrijven."
"Op dezelfde manier trainen we onze deep learning-modellen vooraf op tienduizenden moleculen die algemene eigenschappen hebben die interessant zijn voor ons doel. Zodra de modellen voldoende informatie hebben, 'verfijnen' we ze op een specifiekere set, gericht op wat we willen bereiken, zoals bioactief zijn in een bepaald doeleiwit. En dit blijkt meermaals heel goed te werken, soms met niet meer dan vijf moleculen in de tweede fase van training! In het Science Advances-artikel gebruikten we er 40."
sNELLER BETERE BESLISSINGEN
Uiteindelijk slaagden Grisoni en haar collega's erin 12 nieuwe bioactieve verbindingen te identificeren voor zogenaamde lever X-receptoren. Dit zijn veelbelovende doelwitten voor geneesmiddelen wegens de regulerende rol die ze spelen bij het vetmetabolisme en ontstekingen.
Natuurlijk is er, zoals altijd bij innovatief onderzoek, nog een lange weg te gaan. Zo werd de chemische ruimte in het experiment beperkt tot 17 eenstapsreacties om ervoor te zorgen dat de verbindingen goed aansloten op de on-chip experimenten. Grisoni wijst er ook op dat de structurele diversiteit van de nieuwe bioactieve verbindingen nog vrij beperkt is en misschien nog moet worden uitgebreid.
Toch is de in Italië geboren onderzoeker heel tevreden met de resultaten. "Onze studie laat als eerste zien hoe je AI-taalmodellen voor het ontwerpen van moleculen succesvol kan combineren met geautomatiseerde synthese op een chip. De wereld van medicijnontwikkeling staat voor ongekende mogelijkheden, aangedreven door nieuwe AI-technologie en interdisciplinaire samenwerking op het gebied van moleculair ontwerp en synthese. In de toekomst zullen benaderingen zoals de onze medicinale chemici ondersteunen om sneller 'betere beslissingen' te nemen."
Mediacontact
Meer over AI



Het laatste nieuws


